
Page 64 - Genetics_From_Genes_to_Genomes_6th_FULL_Part3
P. 64
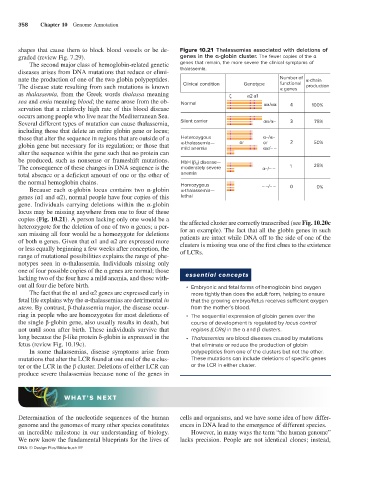
358 Chapter 10 Genome Annotation
shapes that cause them to block blood vessels or be de- Figure 10.21 Thalassemias associated with deletions of
graded (review Fig. 7.29). genes in the α-globin cluster. The fewer copies of the α
The second major class of hemoglobin-related genetic genes that remain, the more severe the clinical symptoms of
diseases arises from DNA mutations that reduce or elimi- thalassemia.
nate the production of one of the two globin polypeptides. Number of -chain
The disease state resulting from such mutations is known Clinical condition Genotype functional production
genes
as thalassemia, from the Greek words thalassa meaning 2 1
sea and emia meaning blood; the name arose from the ob- Normal / 4
servation that a relatively high rate of this blood disease 100%
occurs among people who live near the Mediterranean Sea.
Several different types of mutation can cause thalassemia, Silent carrier / – 3 75%
including those that delete an entire globin gene or locus;
those that alter the sequence in regions that are outside of a Heterozygous –/ –
globin gene but necessary for its regulation; or those that -thalassemia— or or 2 50%
/– –
mild anemia
alter the sequence within the gene such that no protein can
be produced, such as nonsense or frameshift mutations. HbH ( ) disease—
4
The consequence of these changes in DNA sequence is the moderately severe –/– – 1 25%
total absence or a deficient amount of one or the other of anemia
the normal hemoglobin chains. Homozygous
Because each α-globin locus contains two α-globin -thalassemia— – –/– – 0 0%
genes (α1 and α2), normal people have four copies of this lethal
gene. Individuals carrying deletions within the α-globin
locus may be missing anywhere from one to four of these
copies (Fig. 10.21). A person lacking only one would be a the affected cluster are correctly transcribed (see Fig. 10.20c
heterozygote for the deletion of one of two α genes; a per- for an example). The fact that all the globin genes in such
son missing all four would be a homozygote for deletions patients are intact while DNA off to the side of one of the
of both α genes. Given that α1 and α2 are expressed more clusters is missing was one of the first clues to the existence
or less equally beginning a few weeks after conception, the of LCRs.
range of mutational possibilities explains the range of phe-
notypes seen in α-thalassemia. Individuals missing only
one of four possible copies of the α genes are normal; those essential concepts
lacking two of the four have a mild anemia, and those with-
out all four die before birth. • Embryonic and fetal forms of hemoglobin bind oxygen
The fact that the α1 and α2 genes are expressed early in more tightly than does the adult form, helping to ensure
fetal life explains why the α-thalassemias are detrimental in that the growing embryo/fetus receives sufficient oxygen
utero. By contrast, β-thalassemia major, the disease occur- from the mother’s blood.
ring in people who are homozygotes for most deletions of • The sequential expression of globin genes over the
the single β-globin gene, also usually results in death, but course of development is regulated by locus control
not until soon after birth. These individuals survive that regions (LCRs) in the α and β clusters.
long because the β-like protein δ-globin is expressed in the • Thalassemias are blood diseases caused by mutations
fetus (review Fig. 10.19c). that eliminate or reduce the production of globin
In some thalassemias, disease symptoms arise from polypeptides from one of the clusters but not the other.
mutations that alter the LCR found at one end of the α clus- These mutations can include deletions of specific genes
ter or the LCR in the β cluster. Deletions of either LCR can or the LCR in either cluster.
produce severe thalassemias because none of the genes in
WHAT’S NEXT
Determination of the nucleotide sequences of the human cells and organisms, and we have some idea of how differ-
genome and the genomes of many other species constitutes ences in DNA lead to the emergence of different species.
an incredible milestone in our understanding of biology. However, in many ways the term “the human genome”
We now know the fundamental blueprints for the lives of lacks precision. People are not identical clones; instead,
DNA: © Design Pics/Bilderbuch RF